8/2021
Орфанное заболевание



Болезнь черной мочи
Отличительным признаком болезни является выделение мочи, которая быстро темнеет и становится черной при контакте с воздухом.
9/2021
Орфанное заболевание

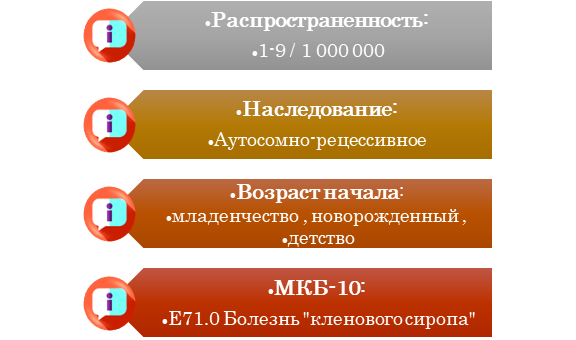
Болезнь «кленового сиропа»
Болезнь «кленового сиропа» (лейциноз, короткоцепочечная кетоацидурия, болезнь разветвленных кислот, разветвленноцепочечная кетонурия) – редкое наследственное нарушение метаболизма, обусловленное дефицитом дегидрогеназного комплекса альфа-кетокислот с разветвленной цепью и последующего накопления метаболитов-предшественников, аминокислот с длинной разветвленной цепью и их альфа-кетокислот. Это мощные нейротоксины, ответственные за быстрое начало отека и диффузную церебральную демиелинизацию. Заболевание впервые было описано в 1954 году [1].
10/2021
Орфанное заболевание

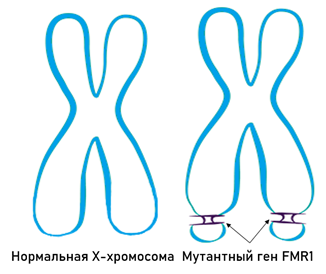
Синдром ломкой X-хромосомы
Синдром ломкой Х-хромосомы, также называемый синдромом Мартина-Белла, является заболеванием с неменделирующим наследованием, наиболее распространенной наследственной причиной легкой и тяжелой умственной отсталости и наиболее частой моногенной причиной расстройств аутистического спектра [1].

1/2022
Орфанное заболевание
Синдром Ангельмана – синдром «ангела»
Синдром Ангельмана (синдром «счастливой куклы» или «счастливой марионетки», Angelman syndrome, СА) — генетическое заболевание, в основе которого лежат морфологические или функциональные нарушения локуса q11-q13 копии материнской хромосомы 15, сопровождающиеся потерей экспрессии гена UBE3A в нейронах головного мозга. Впервые данное заболевание описал английский педиатр Harry Angelman в 1965 году, когда он обобщил фенотипы трех умственно отсталых детей, которые были охарактеризованы как puppet children — «дети-куклы» из-за необычной позиции рук и своеобразных дерганных и отрывистых движений конечностей (с изоляцией отдельных фаз движений и фиксацией промежуточных положений конечностей).

2/2022
Орфанное заболевание
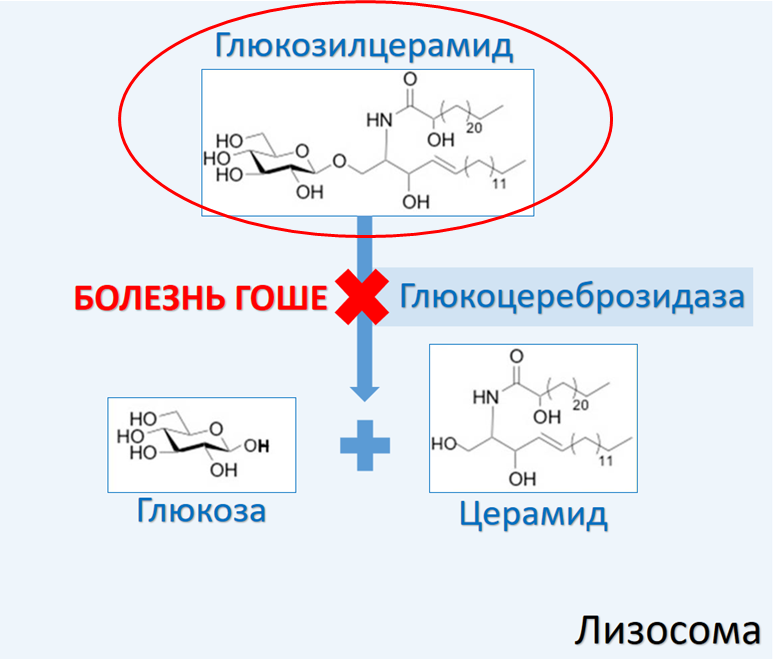
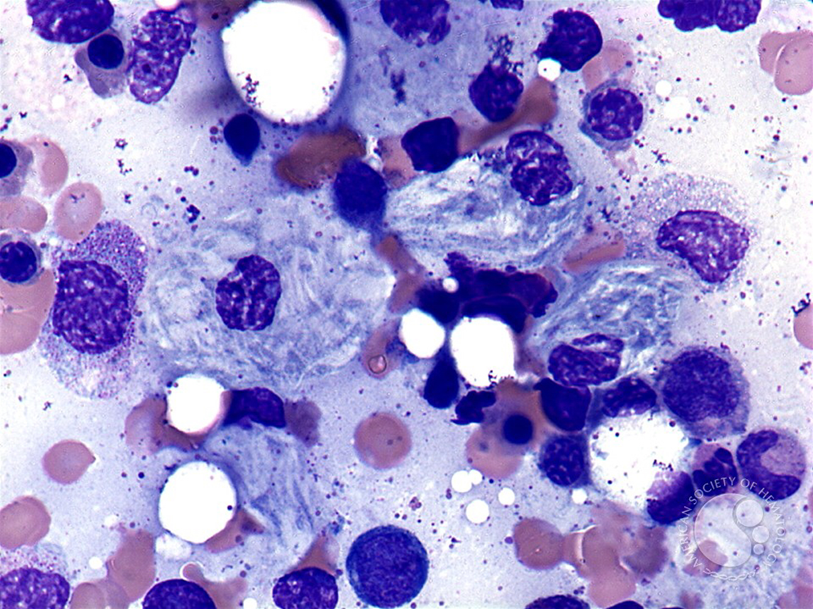
Болезнь Гоше
Лизосомальные болезни накопления представляют собой группу гетерогенных наследственных заболеваний, вызванных мутациями, влияющими на гены, которые кодируют либо функцию лизосомальных ферментов, необходимых для деградации широкого спектра сложных макромолекул, либо функцию специфических транспортеров, необходимых для экспорта деградированных макромолекул из лизосом. Возникающая в результате лизосомная дисфункция приводит к клеточной дисфункции и клиническим аномалиям. В группе сфинголипидозов наблюдается дисфункция ферментативной деградации метаболитов, которые являются важными компонентами клеточных мембран и регуляторами различных сигнальных путей [1].

4/2022
Орфанное заболевание

Х-сцепленный гипофосфатемический рахит
Х-сцепленный гипофосфатемический рахит (ХГР) является редким генетическим заболеванием, для которого характерно снижение реабсорбции фосфора в проксимальных почечных канальцах, что приводит к гипофосфатемии, являющейся причиной дефекта минерализации кости. Данное состояние характеризуется высокой концентрацией фибробластоподобного фактора роста 23 (FGF23). Клиническими проявлениями гипофосфатемии являются сложные деформации скелета, тяжелый рахит, нарушение роста в детском возрасте, у взрослых пациентов к вторичным осложнениям, ухудшающим качество жизни, относятся остеоартрозы крупных и мелких суставов. Несмотря на разработку таргетной терапии, данная группа пациентов нуждается в междисциплинарной специализированной помощи, в связи с множественными сопутствующие заболевания. Основной задачей специалистов является координация пациентов для оптимизации лечения и улучшения качества жизни пациентов.
9-10/2022
Орфанное заболевание

Синдромы множественных эндокринных неоплазий
Редкие (орфанные) заболевания – это патологии, которые встречаются с частотой не более 1 случая на 2 тыс. человек. При отсутствии надлежащего лечения орфанные заболевания являются инвалидизирующими и могут угрожать жизни. Одной из групп орфанных заболеваний являются синдромы множественных эндокринных неоплазий (МЭН), клиническая картина которых индивидуальна и обусловлена нарушением секреции определенных гормонов. В связи с генетической природой МЭН своевременное обнаружение мутаций в локусах генов MEN и RET позволяет вовремя инициировать лечение и предотвратить развитие конкурирующих заболеваний в рамках одного синдрома.

